How I Built A Custom Protein-Binding Dataset for Machine Learning



Table of Contents #
- Table of Contents
- Background: CNNs can predict binding sites
- The Rationale: But wait — does the data really matter that much?
- The Process: Building the PepBDB-ML dataset
- 1. Initial filtering
- 2. Sequence extraction using BioPython
- 3. Binding sequences determination with PRODIGY
- 4. Adding sequence level information with AAindex and PSI-BLAST
- 5. Structural information using BioPython
- Optional: Generating an image dataset
- The Results: Visualizing the PepBDB-ML database
- The Discussion: Key takeaways and learnings
90% of the time I tell people I work with proteins, they assume I’m trying to get jacked.
And okay, fine, they’re right.
But really, I’m talking about biological proteins— incredible molecular machines that make it possible for your body to function.
 on [Unsplash](https://unsplash.com?utm_source=medium&utm_medium=referral)](https://cdn-images-1.medium.com/max/13936/0*ib4VLHvLb-nWJjTA)
Proteins are involved in vital metabolic processes — including getting that summer body — and peptide-protein interactions (PepPIs) are among the most important.
(Peptide = short protein segments).
Recently, I built a dataset I could use to study PepPIs. And by the end of this article, you will have too.
Here’s a quick peek at a few things I discovered in what I’m calling the PepBDB-ML database:
Protein residue pseudo angles take on a bimodal distribution.
Substitution scores for residues are normally distributed (but there is one exception).
Amino acids that are typically found in beta sheets might be more open to swapping with isoleucine than other residues.
But I’m getting ahead of myself —
Let’s start with some background.
Background: CNNs can predict binding sites #
Recently, I read a paper that used CNNs to predict peptide-protein binding sites from a sequence.
They took a context window from the sequence (7 amino acids, AKA residues), enriched it with data on hydrophobicity, exposure, relative angles, etc., and transformed it into an image.
- Repeating this across a body of residues created a massive dataset of training images.
Analogy: Imagine you had a Lego Deathstar, and you took a picture of each brick — and these pictures were force-imbued, so you could see things like the age and weight of each brick. Label each image with data about where it goes in the Deathstar (e.g., interior vs exterior), and you’d learn how to classify new pieces. Protein = Deathstar, Lego brick = amino acid
- Feeding this into a CNN yielded a model with binding site detection rates twice as high as previously published methods.
](https://cdn-images-1.medium.com/max/2000/0*nQ_R3fP2WysCt0Jk.jpg)
I thought this approach was fascinating, but I saw an opportunity for improvement: residue secondary structure tags, angles, and accessible surface area were all predicted using SPIDER2 — but what if I just got the actual data from experimental structures instead?
That’s precisely what I did using the PepBDB, a structural database of 3D peptide-receptor complexes (all structures are curated from the RCSB PDB, which provides data under license CC0).
](https://cdn-images-1.medium.com/max/4660/1*PBEv5SFgjEAvhMI2wyodRQ.png)
By using all the information applied in Visual, along with some structure-specific features, I could:
Expand the dataset
Potentially improve model performance
Save the world
(Two truths, one lie).
So that is precisely what I set out to do with PepBDB-ML
The Rationale: But wait — does the data really matter that much? #
Well, there’s one key reason I think focusing on data is one of the highest-leverage activities in AI: data is our greatest constraint.
Sure, compute is critical — but the data hydra has three heads:
1. Availability #
Heard about the recent content licensing deal between OpenAI and News Corp, Vox Media, and The Atlantic? The agreement with News Corp alone is believed to be worth over $250 million.
While a lot of AI training data is either unprotected or open-source, the good stuff is often unavailable, expensive, or hard to find. Which leads to the next head…
2. Quality #
Let’s use an example.
If LLMs right now are really good at generating text, why not use them to create more training data? Unfortunately, making AI eat its own tail causes something called Model Autophagy Disorder, or MAD — and yes, they came up with the acronym before deciding what it stood for.
Bottom line? Synthetic data can only take us so far. In the world of biology, that means we need reliable, experimental data (like the structures from PepBDB).
3. Interpretability #
In biology specifically, human-readable data is often the most helpful. It allows us to unpack machine learning models and see things like:
Critical features.
Relative importance.
The future (I’m only half-joking).
For example, when it comes to peptide-protein binding, what’s more impactful — hydrophobicity or half-sphere exposure? Without labels, we can’t answer that question.
Documented data means that models and humans can both learn more about biology.
Key Points:
We can always fine-tune ML/DL models, but we’re bottlenecked by the quality of our data.
Availability, quality, and interpretability are crucial aspects of biological datasets.
Now that you’re convinced we’re not wasting time, let’s get into the meat of this project: building the dataset.
The Process: Building the PepBDB-ML dataset #
Before we begin, let’s decide what we want our dataset to look like.
A tabular version, where each row represents a residue from a protein complex and each column is a feature (like residue hydrophobicity, polarizability, secondary structure within the protein). These features will be the ones used in Visual, along with some extras.
An image version, where each image is essentially 7 adjacent rows from the tabular dataset, representing a context window for the central amino acids.
With this plan in mind, we can focus our efforts on the tabular dataset. Since the PepBDB database contains structures, we can use structural and sequence information. Here’s how we’ll approach this:
Initial filtering
Sequence extraction + filtering
Binding residue determination with Prodigy
Sequence level information (AAindex, PSI-BLAST)
Structural information from BioPython (DSSP, HSE, Pseudo-angles)
There’s going to be a lot of code coming up, so buckle up.
Before we begin, we’ll start with our imports:
# gendata.py
import pandas as pd
import numpy as np
import argparse
import os
import pickle
from aaindex import *
from helpers import *
from paths import *
What are those ones at the bottom? Don’t worry — we’ll get to those soon.
(Heads up — since we’re going to be jumping between a few scripts, I’ll include the filename at the top of each code block).
1. Initial filtering #
The PepBDB offers the peptidelist.txt file as part of its download. This file provides information about each peptide-protein pair in the repository.
# gendata.py
## Load the peptidelist.txt file
peptide_list = pd.read_csv(peptide_list_txt, sep='\\\\s+', header=None)
headers = ['PDB ID', 'Peptide Chain ID', 'Peptide Length', 'Number of Atoms in Peptide',
'Protein Chain ID', 'Number of Atoms in Protein',
'Number of Atom Contacts', 'unknown1', 'unknown2', 'Resolution', 'Molecular Type']
peptide_list.columns = headers
# 'pepbdb' below is the path to the PepBDB directory, imported from `paths`
peptide_list['Peptide Path'] = pepbdb + peptide_list['PDB ID'] + '_' + peptide_list['Peptide Chain ID'] + '/peptide.pdb'
peptide_list['Protein Path'] = pepbdb + peptide_list['PDB ID'] + '_' + peptide_list['Peptide Chain ID'] + '/receptor.pdb'
peptide_list.reset_index(drop=True)
## Filter out nucleic acids, low-resolution, and small peptides
peptide_list = peptide_list[peptide_list['Molecular Type'] != 'prot-nuc']
peptide_list = peptide_list[peptide_list['Resolution'] < 2.5]
peptide_list = peptide_list[peptide_list['Peptide Length'] >= 10]
print('\\033[1mInitial filtering done.\\033[0m')
We’ll do some very simple filtering to begin:
Drop all prot-nuc models. These are models that contain a nucleic acid. We’re restricting this dataset to protein-only for the sake of simplicity.
Drop models with resolution higher than 2.5 Å. We want our input data to be high-quality, so let’s drop any low-res models.
Drop models with extra short peptides. This is mainly for the sake of computational efficiency. We’ll drop any peptides shorter than ten amino acids.
In the next step, we’ll use BioPython to extract the sequences from each of our remaining models. Sequence extraction can take some time, which is why I’ve chosen to do it after some initial cleanup.
2. Sequence extraction using BioPython #
PepBDB provides 3D models but not direct sequences. Fortunately, these are relatively simple to extract using BioPython.
To keep the code clean, I’ll create a helper function in a separate file called helpers.py:
# helpers.py
def extract_sequence(pdb_filename: str) -> str:
'''
Helper function that extracts the sequence from a PDB file.
'''
parser = PDBParser()
structure = parser.get_structure('X', pdb_filename)
sequence = ''
contains_unk = False
for model in structure:
for chain in model:
residues = chain.get_residues()
for residue in residues:
if residue.get_resname() == 'HOH': # ignoring water
continue
if residue.get_resname() == 'UNK':
sequence += 'X' # add X for each 'UNK' residue
else:
sequence += seq1(residue.get_resname()) # seq1 converts 3-letter code to 1-letter code
return sequence
Then, I’ll import it into our data generation script and apply it to our dataset:
# gendata.py
peptide_list['Peptide Sequence'] = peptide_list['Peptide Path'].apply(extract_sequence)
peptide_list['Protein Sequence'] = peptide_list['Protein Path'].apply(extract_sequence)
print('\\\\033[1mSequences extracted.\\\\033[0m')
Now that we have sequences for each PDB structure, let’s remove the problematic ones (sequences with non-standard amino acids):
# gendata.py
peptide_list = peptide_list[~peptide_list['Peptide Sequence'].str.contains('[UOBZJX\\\\\\\\*]', regex=True)]
peptide_list = peptide_list[~peptide_list['Protein Sequence'].str.contains('[UOBZJX\\\\\\\\*]', regex=True)]
peptide_list = peptide_list.drop_duplicates(subset=['Peptide Sequence', 'Protein Sequence'])

3. Binding sequences determination with PRODIGY #
PRODIGY (PROtein binDIng enerGY prediction) is a tool for predicting binding affinities for protein structures from 3D data.
Part of that involves identifying residues in contact with each other — i.e., involved in binding.
I used PRODIGY on every PDB in the PepBDB leftover from the last preprocessing step. The program outputs a .icfile — a simple text file that lists the residues involved in binding.
Here’s an example for calcium-bound cyclin (1A03):
GLY 78 A GLN 71 B
ILE 13 A TYR 84 B
LYS 40 A MET 1 B
GLU 86 A HIS 27 B
LEU 88 A LEU 12 B
ALA 8 A ILE 15 B
MET 1 A LYS 40 B
ALA 87 A ILE 13 B
PHE 70 A LEU 77 B
GLN 71 A MET 82 B
ALA 8 A ALA 8 B
ILE 74 A LEU 77 B
LYS 89 A LYS 26 B
Processing this data gives us two lists of indices, each referring to a binding residue.
The peptides and receptors in each row are available as distinct PDBs, i.e. their models contain one chain each. For our purposes, PRODIGY needs a complex (2 chains), so we’ll make a temporary file and process the output:
# helpers.py
def label_residues(peptide_path: str, protein_path: str) -> List:
# creating a temporary file with the peptide and protein
with tempfile.NamedTemporaryFile(suffix='.pdb', delete=False) as temp_file:
output_path = temp_file.name
subprocess.run(['cat', peptide_path, protein_path], stdout=temp_file)
# ensuring the temp file is properly closed before using it in subprocess
temp_file.close()
# obtaining binding residues using PRODIGY
try:
subprocess.run(['prodigy', '-q', '--contact_list', output_path], check=True)
except subprocess.CalledProcessError as e:
print(f"An error occurred while processing the files: {peptide_path} & {protein_path}")
print(f"Error: {e}")
# the .ic file will have the same root name as the input file
ic_file_path = output_path.replace('.pdb', '.ic')
# check if the .ic file exists before reading
if not os.path.exists(ic_file_path):
raise FileNotFoundError(f"{ic_file_path} not found after running PRODIGY.")
# are you actually reading these comments? kudos! if yes — reply with a 😼
contacts = pd.read_csv(ic_file_path, sep='\s+', header=None)
contacts.columns = ['peptide_residue', 'peptide_index', 'peptide_chain', 'protein_residue', 'protein_index', 'protein_chain']
peptide_binding_residues = sorted(list(contacts['peptide_index'].unique()))
protein_binding_residues = sorted(list(contacts['protein_index'].unique()))
os.remove(output_path)
return peptide_binding_residues, protein_binding_residues
Now, let’s apply it to our dataset:
# gendata.py
print('Running PRODIGY to determine binding contacts...')
## determine binding residues from peptide-protein complexes
peptide_list['Peptide Binding Indices'] = np.nan
peptide_list['Protein Binding Indices'] = np.nan
for index, row in peptide_list.iterrows():
peptide_path = row['Peptide Path']
protein_path = row['Protein Path']
peptide_binding_positions, protein_binding_positions = label_residues(peptide_path, protein_path)
peptide_list.at[index, 'Peptide Binding Indices'] = str(peptide_binding_positions)
peptide_list.at[index, 'Protein Binding Indices'] = str(protein_binding_positions)
print('\\\\033[1mBinding indices identified.\\\\033[0m')
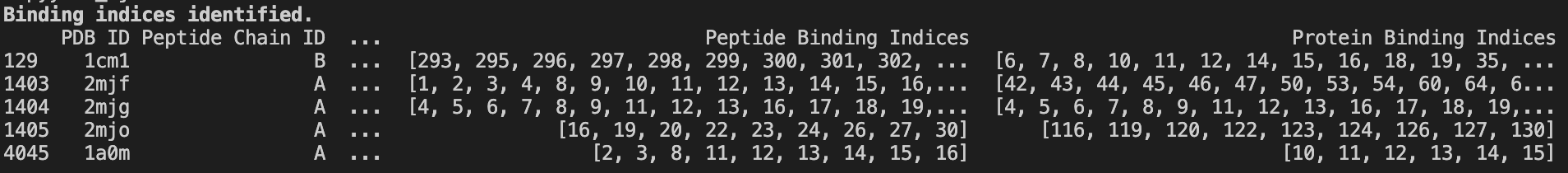
The Biology: You might be wondering — why build this dataset if we can use PRODIGY to get binding sites? Well, PRODIGY generally needs two chains in complex. Our dataset is aimed at helping ML/DL models predict binding sites with just one model chain.
4. Adding sequence level information with AAindex and PSI-BLAST #
AAindex #
We’ve now got a list of protein sequences. We can enrich our dataset by adding publically available, residue-specific information from the Amino Acid Index Database — a cornucopia of known, reference amino acid features.
For example, here’s hydrophobicity:
H ARGP820101
D Hydrophobicity index (Argos et al., 1982)
R PMID:7151796
A Argos, P., Rao, J.K.M. and Hargrave, P.A.
T Structural prediction of membrane-bound proteins
J Eur. J. Biochem. 128, 565-575 (1982)
C JOND750101 1.000 SIMZ760101 0.967 GOLD730101 0.936
TAKK010101 0.906 MEEJ810101 0.891 ROSM880104 0.872
CIDH920105 0.867 LEVM760106 0.865 CIDH920102 0.862
MEEJ800102 0.855 MEEJ810102 0.853 ZHOH040101 0.841
CIDH920103 0.827 PLIV810101 0.820 CIDH920104 0.819
LEVM760107 0.806 NOZY710101 0.800 GUYH850103 -0.808
PARJ860101 -0.835 WOLS870101 -0.838 BULH740101 -0.854
I A/L R/K N/M D/F C/P Q/S E/T G/W H/Y I/V
0.61 0.60 0.06 0.46 1.07 0. 0.47 0.07 0.61 2.22
1.53 1.15 1.18 2.02 1.95 0.05 0.05 2.65 1.88 1.32
//
We’re interested in those values along the bottom. These are the feature Infinity Stones I collected for our dataset Gauntlet:
- Hydrophobicity: a measure of the tendency of that side chain to be located in the interior of a native protein molecule. Normalized to be unitless.
Note: In the original paper, the units were ΔFₜ — for the transfer of the amino acid side chain from organic solvent to water at 25° in cal/mole. Here, they are all made positive and normalized to a value of 1.0, which means they adjusted the scale so that the average value is 1.0.
Steric parameter: the tendency of amino acids to be found at the surface of globular proteins as a function of side-chain structure. Unitless, but calculated based on van der Waals radii.
Residue volume: The space a residue occupies. Units are Ų.
Polarizability: a measure of how easily an amino acid’s electron cloud will form dipoles in response to external forces. Unitless, a relative measure.
Average relative probability of helix: the relative helix residue probability in 47 proteins. Unitless, relative measure.
Average relative probability of beta sheet: the relative beta sheet residue probability in 47 proteins. It is also a unitless, relative measure.
- Isoelectric point: the pH at which an amino acid is electrically neutral. Units are pH.
The Biology: Why these features? Here’s an example: we might expect binding residues to be on the surface of the protein, where it can easily bind to other peptides — so a measure like steric parameter is useful.
Using AAindex1, I load all of these indices into a script aaindex.py full of dictionaries and a function to access them:
# aaindex.py
def feature_vector(seq: str, feature_type: dict):
'''
Returns a list of feature values corresponding to each
residue in sequence, according to AAindex1.
Feature types include:
- Hydrophobicity
- Steric parameter
- Residue volume
- Polarizability
- Average relative probability of helix
- Average relative probability of beta
- Isoelectric point
'''
return [feature_type[aa] for aa in seq]
hydrophobicity = {
'A': 0.61,
...,
'V': 1.32
}
Next, I extract this information from every sequence:
# gendata.py
print('Adding residue-level data from AAindex1...')
## Add rich data from AAindex1
features = {
'Hydrophobicity': hydrophobicity,
'Steric Parameter': steric_parameter,
'Volume': residue_volume,
'Polarizability': polarizability,
'Helix Probability': average_relative_probability_of_helix,
'Beta Probability': average_relative_probability_of_beta_sheet,
'Isoelectric Point': isoelectric_point
}
for feature_name, feature_function in features.items():
peptide_list[f'Peptide {feature_name}'] = peptide_list['Peptide Sequence'].apply(lambda x: feature_vector(x, feature_function))
peptide_list[f'Protein {feature_name}'] = peptide_list['Protein Sequence'].apply(lambda x: feature_vector(x, feature_function))
print('\\033[1mAAindex features added.\\033[0m')
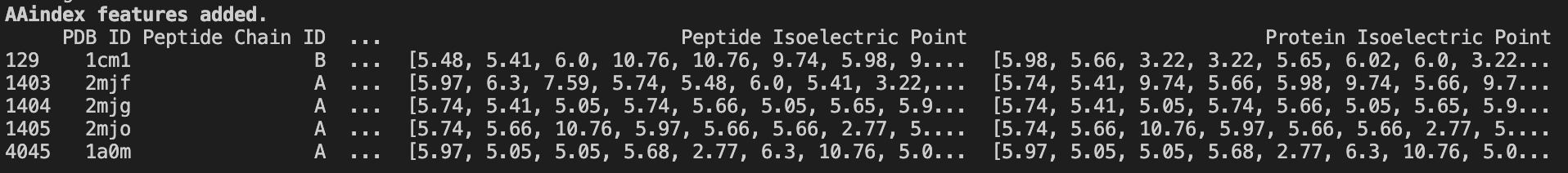
Note: since completing this piece, I’ve discovered a Python package for accessing all data from the AAindex. Which means I didn’t need to write this script from scratch. Yes, I have feelings about this. No, I will not be discussing them.
PSI-BLAST #
Next, let’s use PSI-BLAST to get residue-level conservation scores.
PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool) is a sequence alignment tool that lets us build a Position Specific Scoring Matrix (PSSM) from a sequence.
A PSSM tells us how conserved a given residue is at a given position in the sequence. High score = highly conserved.
The Biology: Why do we want this information? Binding motifs are typically highly conserved — alterations could mean loss of binding function, stymying organism survival.
Here’s an example for the same protein we looked at earlier, 1A03:
Last position-specific scoring matrix computed, weighted observed percentages rounded down, information per position, and relative weight of gapless real matches to pseudocounts
A R N D C Q E G H I L K M F P S T W Y V A R N D C Q E G H I L K M F P S T W Y V
1 M -3 -4 -4 -5 -4 -3 -4 -5 -4 -1 0 -4 10 -2 -5 -4 -3 -4 -3 -1 0 0 0 0 0 0 0 0 0 0 0 0 100 0 0 0 0 0 0 0 2.17 0.45
2 A 1 -3 0 -2 1 -2 -1 1 -3 -3 -2 -2 -3 -4 6 0 -1 -4 -4 -3 14 0 5 0 4 0 3 12 0 0 5 0 0 0 48 7 2 0 0 0 0.92 0.21
3 S -1 0 -1 -2 1 -2 -2 -2 -2 -1 -3 -1 -2 -3 -2 4 4 -4 -3 -2 2 6 0 1 3 0 1 1 0 4 1 2 0 0 0 40 37 0 0 1 0.61 0.25
4 P -2 -2 -2 3 -5 4 3 -3 -2 -4 -3 1 -3 -5 4 -1 -2 -5 -4 -4 0 0 0 15 0 23 22 0 0 0 3 9 0 0 25 1 0 0 0 0 0.80 0.28
....
86 E -2 -1 0 3 -4 0 4 -3 2 -3 -2 3 -3 -4 0 -2 -2 -4 -1 -3 0 0 3 20 0 0 33 0 5 1 5 26 0 0 5 0 0 0 2 0 0.55 0.20
87 A -1 -1 0 0 -3 2 0 -3 2 -1 -2 2 -2 3 -3 0 -1 -1 3 -1 4 0 3 6 0 11 5 0 6 4 0 17 0 14 0 9 0 0 15 6 0.21 0.12
88 L -2 -3 -3 -4 -3 -3 -4 -4 -3 1 3 -3 3 5 -4 1 -1 -1 2 -1 0 0 0 0 0 0 0 0 0 7 28 0 9 34 0 14 2 0 6 0 0.54 0.19
89 K -3 0 -1 0 -5 2 2 -3 9 -5 -4 2 -3 -3 -3 -2 -3 -4 0 -4 0 2 0 5 0 7 11 0 62 0 0 12 0 0 0 0 0 0 0 0 1.18 0.25
90 G 0 -2 3 -1 -3 -2 -2 5 -2 -4 -4 -2 -3 -4 -3 0 -2 -3 -3 -3 6 0 23 0 0 0 0 72 0 0 0 0 0 0 0 0 0 0 0 0 0.87 0.08
K Lambda
Standard Ungapped 0.1364 0.3155
Standard Gapped 0.0519 0.2670
PSI Ungapped 0.1697 0.3169
PSI Gapped 0.0519 0.2670
NCBI provides PSI-BLAST as a command-line tool, so we’ll write some code that invokes it for all of our sequences.
We’ll create a temporary FASTA file with the sequence so we can input it into PSI-BLAST.
swissprot, which is a path to the SwissProt database (imported from paths, licensed under CC BY 4.0, and can be downloaded from NCBI. You can use other databases, but this one was smaller and easier to manage.
# helpers.py def get_pssm_profile(sequence: str) -> pd.DataFrame: ''' Uses blast+ psiblast to generate PSSM profile from a temporary fasta file. ''' with tempfile.NamedTemporaryFile(suffix='.fa', delete=False) as fasta_file: fasta_file.write(f'>tmp\n{sequence}'.encode('utf-8')) fasta_path = fasta_file.name pssm_fd, pssm_path = tempfile.mkstemp(suffix='.pssm') os.close(pssm_fd) try: subprocess.run( ['psiblast', '-query', fasta_path, '-db', swissprot, '-num_iterations', '3', '-evalue', '0.001', '-out_ascii_pssm', pssm_path], stdout=subprocess.DEVNULL, stderr=subprocess.DEVNULL, check=True ) with open(pssm_path, 'r') as file: lines = file.readlines() # Skip the header lines and parse the matrix pssm_data = [] for line in lines[3:len(sequence) + 3]: parts = line.strip().split() scores = parts[1:22] # First 20 columns are scores for each amino acid pssm_data.append(scores) columns = ['AA'] + list('ARNDCQEGHILKMFPSTWYV') df_pssm = pd.DataFrame(pssm_data, columns=columns) finally: os.remove(fasta_path) os.remove(pssm_path) return df_pssm.TNow, let’s apply this function to our dataset:
# gendata.py print('Now generating and filtering PSSMs...') ## Add PSSM profiles into dataframe peptide_list['Peptide PSSM'] = peptide_list['Peptide Sequence'].apply(get_pssm_profile) peptide_list['Protein PSSM'] = peptide_list['Protein Sequence'].apply(get_pssm_profile) print('\\033[1mPSSMs generated.\\033[0m')
5. Structural information using BioPython #
Now that we’ve extracted our information from the sequences, let’s take a closer look at the 3D models.
Secondary structure codes, half-sphere exposure, and from DSSP #
The Dictionary of Secondary Structure in Proteins (DSSP) determines the most likely secondary structure assignment of a residue given a 3D structure. While the original paper uses SPIDER2 to predict this, we can use the PDB itself.
# helpers.py
def hse_and_dssp(pdb_file: str):
'''
Get the HSE and DSSP codes of a PDB's residues.
'''
with warnings.catch_warnings():
warnings.simplefilter(action='ignore', category=FutureWarning)
# Initialize PDBParser
pdb_parser = PDBParser()
structure = pdb_parser.get_structure('structure', pdb_file)
hse = HSExposure.HSExposureCA(structure)
dssp = DSSP(structure[0], pdb_file)
# Half-sphere exposure values
hse_up, hse_down, pseudo_angle = zip(*[(res[1][0], res[1][1], res[1][2]) for res in hse.property_list])
# Extract DSSP values into separate lists
ss, asa, phi, psi = zip(*[(res[2], res[3], res[4], res[5]) for res in dssp.property_list])
print(f'\\rData acquired for {pdb_file}')
return hse_up, hse_down, pseudo_angle, ss, asa, phi, psi
def safe_hse_and_dssp(pdb_file):
'''
Helper function to handle errors and apply `hse_and_dssp`.
'''
# Empty list to store error-producing filenames
error_files = []
try:
return hse_and_dssp(pdb_file)
except Exception as e:
print(f'Error processing file: {pdb_file} - {e}')
error_files.append(pdb_file)
return [None] * 7 # Return a list of None values to match the expected output structure
Let’s break down each of these features:
Secondary structure
ss: These are DSSP structure assignment codes.Relative accessible surface area
asa: a measure of how accessible a residue in the sequence is by its surrounding solvent, normalized by the maximum possible ASA for that residue. (Unitless, but the maximum ASA is in Ų)phiangles: the angle of rotation about the (nitrogen)-(alpha carbon) bond. (In degrees).psiangles: the angle of rotation about the (alpha carbon)-(carbonyl carbon) atom. (In degrees).Half-sphere exposure, up and down
hse_up,hse_down: measures of how buried a residue is in the overall protein structure using the number of Cɑs in the up- and down-facing hemispheres of the amino acid. Units are integer counts.Pseudo angles
pseudo_angle: When calculating HSE, we also get the angle between three adjacent Cɑs. This pseudo-angle helps divide the amino acid into hemispheres and can tell us the shape of the protein backbone at a particular residue. The unit is in degrees.
# gendata.py
print('Adding HSE, ASA, and DSSP codes...')
## Add HSE, ASA, DSSP codes, etc.
peptide_list[['Peptide HSE Up', 'Peptide HSE Down', 'Peptide Pseudo Angles', 'Peptide SS', 'Peptide ASA', 'Peptide Phi', 'Peptide Psi']] = peptide_list['Peptide Path'].apply(lambda x: pd.Series(safe_hse_and_dssp(x)))
peptide_list[['Protein HSE Up', 'Protein HSE Down', 'Protein Pseudo Angles', 'Protein SS', 'Protein ASA', 'Protein Phi', 'Protein Psi']] = peptide_list['Protein Path'].apply(lambda x: pd.Series(safe_hse_and_dssp(x)))
Before we wrap up, there are a few more housekeeping steps we need to take care of:
# One-hot encoding function
def one_hot_encode_array(ss_array):
ss_array = literal_eval(str(ss_array))
length = len(ss_array)
encoding = {code: [0] * length for code in dssp_codes}
for i, code in enumerate(ss_array):
encoding[code][i] = 1
return encoding
def one_hot_encode_row(row):
pep_encoded = one_hot_encode_array(row['Peptide SS'])
prot_encoded = one_hot_encode_array(row['Protein SS'])
new_data = {}
for code in dssp_codes:
new_data[f'Peptide SS {code}'] = pep_encoded[code]
new_data[f'Protein SS {code}'] = prot_encoded[code]
return pd.Series(new_data)
def extend_hse(hse: str) -> str:
"""
Extends a HSE to the full length of the peptide.
"""
hse = list(literal_eval(str(hse)))
hse = [hse[0]] + hse + [hse[-1]]
return hse
## One-hot encode SS codes
ss_columns = peptide_list.apply(one_hot_encode_row, axis=1)
peptide_list = pd.concat([peptide_list, ss_columns], axis=1)
print('\\033[1mHSE, ASA, and DSSP codes added and encoded.\\033[0m')
## Extend HSE and Pseudo Angles to match peptides length. Achieved by duplicating terminal values.
peptide_list['Protein HSE Up'] = peptide_list['Protein HSE Up'].apply(extend_hse)
peptide_list['Protein HSE Down'] = peptide_list['Protein HSE Down'].apply(extend_hse)
peptide_list['Protein Pseudo Angles'] = peptide_list['Protein Pseudo Angles'].apply(extend_hse)
peptide_list['Peptide HSE Up'] = peptide_list['Peptide HSE Up'].apply(extend_hse)
peptide_list['Peptide HSE Down'] = peptide_list['Peptide HSE Down'].apply(extend_hse)
peptide_list['Peptide Pseudo Angles'] = peptide_list['Peptide Pseudo Angles'].apply(extend_hse)
print('\\033[1mHSE data extended.\\033[0m')
- Since this data is going to be used for ML/DL projects, we’ll save ourselves some work by one-hot encoding the secondary structure columns.
- HSEup, HSEdown, and pseudo angles all need three residues, so the terminal residues are left without any values. We’ll shortcut around this — extending the terminal edges by duplicating the first and last values
Finally, we’ll reduce the table so proteins and peptides are no longer sharing rows before converting the whole dataset into a tabular array matching our initial specifications:
# gendata.py
## Reduce to one peptide/protein per row
peptide_cols = [col for col in peptide_list.columns if 'Peptide' in col] # Columns we want to keep in pep_data
peptide_cols.remove('Peptide Length')
protein_cols = [col for col in peptide_list.columns if 'Protein' in col] # Columns we want to keep in pro_data
pep_data = peptide_list[peptide_cols] # Select the peptide data
pro_data = peptide_list[protein_cols] # Select the protein data
# Combine the two dataframes
combined_data = pd.DataFrame(np.vstack([pep_data, pro_data.values]))
combined_data.columns = pro_data.columns
# Replace the original peptide_list with the combined data
peptide_list = combined_data
Next, we’ll define our table-making function:
# helpers.py
def make_tabular_dataset(row: pd.Series) -> pd.DataFrame:
'''
`make_tabular_dataset` is designed to be applied to a row of the input DataFrame
to create a feature array.
'''
feature_dict = row.to_dict()
# get sequence and PSSM
sequence = feature_dict[f'Protein Sequence']
pssm = feature_dict[f'Protein PSSM']
# remove the first row of the PSSM, which is the sequence
pssm = pssm.iloc[1:]
pssm = pssm.values
# get the binding indices list
binding_indices_dummy = [0] * len(sequence)
binding_indices = feature_dict[f'Protein Binding Indices']
for i in range(len(sequence)):
if i+1 in literal_eval(binding_indices):
binding_indices_dummy[i] = 1
# add the sequence as the first element of the array
arr = []
arr.append([char for char in sequence])
# remove these columns from the dictionary; all are either unneeded or have been processed
remove = ['Unnamed: 0', 'PDB ID',
'Number of Atoms in Protein', 'Protein Chain ID', 'Protein Path',
'Number of Atom Contacts', 'Resolution',
'Molecular Type', 'Protein Path', 'Protein Sequence',
'Protein Binding Indices', 'Protein SS', 'Protein PSSM']
# many of the features are stored as strings, so we need to convert them to lists
use_feature_dict = {}
for k, v in feature_dict.items():
if k not in remove:
try:
use_feature_dict[k] = literal_eval(str(v))
except (ValueError, SyntaxError) as e:
print(f"Error parsing key '{k}' with value '{v}': {e}")
# stack all the features
for _, value in use_feature_dict.items():
value_array = [residue_value for residue_value in value]
arr.append(value_array) # add the feature to the array
for aa_row in pssm:
arr.append(aa_row)
arr.append(binding_indices_dummy)
# transpose the array and add the column names
arr = pd.DataFrame(arr).T
columns = ['AA'] + list(use_feature_dict.keys()) + list('ARNDCQEGHILKMFPSTWYV') + ['Binding Indices']
if len(arr.columns) == len(columns):
arr.columns = columns
else:
print("Column length mismatch")
print("Arr shape:", arr.shape)
print("Columns length:", len(columns))
return pd.DataFrame(arr)
# gendata.py
## Create tabular dataset, 1 row per residue
list_of_feature_arrays = []
for i, row in peptide_list.iterrows():
arr = make_tabular_dataset(row)
list_of_feature_arrays.append(arr)
print(f'\\r{i+1}/{peptide_list.shape[0]}', end='')
print('\\033[1m\\nConverted.\\033[0m')
export = pd.concat(list_of_feature_arrays)
export = export.dropna()
export = export.reset_index(drop=True)
export.to_csv(peppi_data_csv, index=False)
But let’s not forget — we also wanted to generate an image dataset. Here’s how we do that with our tabular dataset.
Optional: Generating an image dataset #
We need to write functions that:
Generates a sliding window over each sequence, seven residues wide.
Turns those windows into labeled images.
# helpers.py
def window_maker(sequence_array: pd.DataFrame) -> List[pd.DataFrame]:
'''
Takes a sequence array and returns a list windowed arrays by sliding a window over the input feature array.
The window is centered on the residue of interest.
'''
k = 7 # window size
windows = []
sequence_array = sequence_array.T.reset_index(drop=True)
sequence_length = sequence_array.shape[1]
for i in range(sequence_length):
# N-terminal case
if i < 3:
right = sequence_array.iloc[:, 0:i+4]
if i == 0:
left = right.iloc[:, :-4:-1]
elif i == 1:
left = right.iloc[:, 1::-1]
elif i == 2:
left = right.iloc[:, 1]
window = pd.concat([left, right], axis = 1)
# C-terminal case
elif i >= sequence_length - 3:
left = sequence_array.iloc[:, i-3:]
if i == sequence_length - 3:
right = sequence_array.iloc[:, i+1]
elif i == sequence_length - 2:
right = sequence_array.iloc[:, i-2:i].iloc[:, ::-1]
elif i == sequence_length - 1:
right = sequence_array.iloc[:, i-3:i].iloc[:, ::-1]
window = pd.concat([left, right], axis = 1)
# Standard case
else:
window = sequence_array.iloc[:, i-3:i+4]
windows.append(window)
return windows
def create_images(window: pd.DataFrame, name: str):
'''
Helper function that accepts a window DataFrame and converts it
into a CNN-friendly image.
'''
window = window.T.reset_index(drop=True) # transpose the array and reset the index
window = window.apply(pd.to_numeric, errors='coerce') # convert to numeric
window = window.to_numpy() # convert to numpy array
window_normalized = window * 255 # scale to 0-255
window_uint8 = window_normalized.astype('uint8')
img = Image.fromarray(window_uint8)
img.save(name)
print(f'\\rCreated image: {name}.', end='')
def process_images(list_of_feature_arrays: List[pd.DataFrame], binding_path: str, nonbinding_path: str):
'''
Takes a list sequence feature arrays and uses create_images to turn valid ones into images
in the appropriate folder.
'''
os.makedirs(binding_path, exist_ok=True)
os.makedirs(nonbinding_path, exist_ok=True)
name_index = 0
for arr in list_of_feature_arrays:
residues = arr.pop('AA')
binding_indices = arr.pop('Binding Indices')
binding_indices = binding_indices.to_list()
windows = window_maker(arr)
for i, window in enumerate(windows):
if not window.isna().any().any():
name_index += 1
folder = binding_path if binding_indices[i] == 1 else nonbinding_path
name = f'{folder}/{name_index}.jpg'
create_images(window, name)
else:
continue
print('\\n')
There’s a lot to unpack here, so let’s take it function by function:
window_maker : notice how each element in list_of_feature_arrays is a dataframe corresponding to one particular sequence? window_maker runs a sliding window over it to generate residue-specific arrays.
create_images: this function accepts those windows as input, normalizes the values, and converts them into images.
process_images: here, we run the earlier two functions and save the images to the appropriate folder based on the label. ```python
gendata.py #
Optional step to create images: #
if args.images: print(‘\033[1m\nNow creating images…\033[0m’) process_images( list_of_feature_arrays, binding_path=args.binding_path, nonbinding_path=args.nonbinding_path ) ``` And there we have it!
800,000+ tabular records of rich, residue-level data.
800,000+ images of residues ready for classifier training.
Encoded and transformed to be ML/DL friendly.
Behold — the PepBDB-ML database.
For the data-curious among us — don’t worry. Here are some visualizations to explore the dataset now that we have it available:
The Results: Visualizing the PepBDB-ML database #
Amino acid distributions #
| 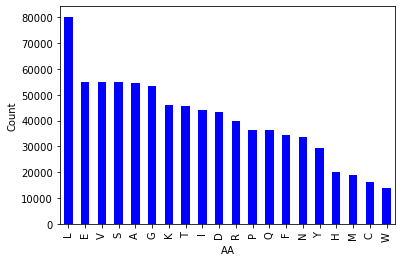 |
Figure 1. Bar chart of amino acid counts in PepBDB-ML.
There isn’t too much to note here, apart from the curious observation that Leucine outnumbers every other amino acid in the database.
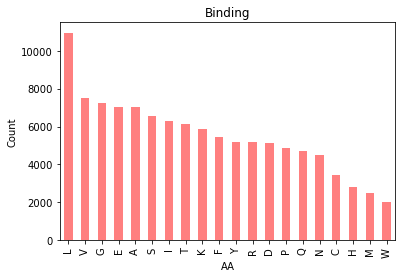
| 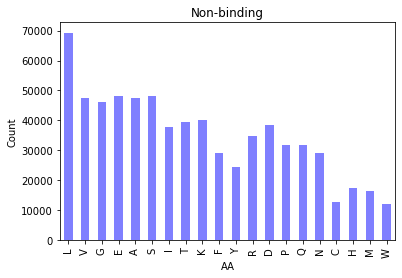 |
Figure 2. Bar chart of amino acid distributions in binding (left) and non-binding residues (right).
Splitting our amino acid counts by binding vs. nonbinding residues shows us the relative proportion of AAs differs ever so slightly. Here’s a view of both in the same plot:
|  |
Figure 3. Bar chart of amino acid percentages by class in PepBDB-ML.
Takeaways:
The residue makeup of binding and nonbinding residues might differ slightly.
(In other breaking news, the sky is blue).
Correlation mapping #
|  |
Figure 4. Correlation heatmap of features in PepBDB-ML.
A correlation heatmap helps us visualize correlations between variables in our dataset. The colours and labels tell us the strength and directions of those correlations.
Some takeaways:
A strong relationship between residue polarizability and volume (0.96). This makes a lot of sense — large amino acids have more electrons around them, which makes them more polarizable.
A weak relationship between psi angle and the residue being involved in a helix. Psi angles fall between -50–100° in helices, which may explain part of what we see here.
An interesting point seems to be a weakly positive relationship between relative beta-sheet folding probability and substitutability with Isoleucine.
Feature distributions #
|  |
Figure 5. Distributions of 41 features found in PepBDB-ML.
Some interesting callouts:
Pseudoangles have a bimodal distribution.
PSI-BLAST substitution scores are primarily normally distributed, but…
Tryptophan (W) skews low — meaning most sequences don’t want to change partners with W. Since tryptophan is the largest amino acid (204.23 g/mol), its bulk can cause steric hindrance should it be swapped with smaller residues.
Now, let’s compare distributions between binding and non-binding residues:
| 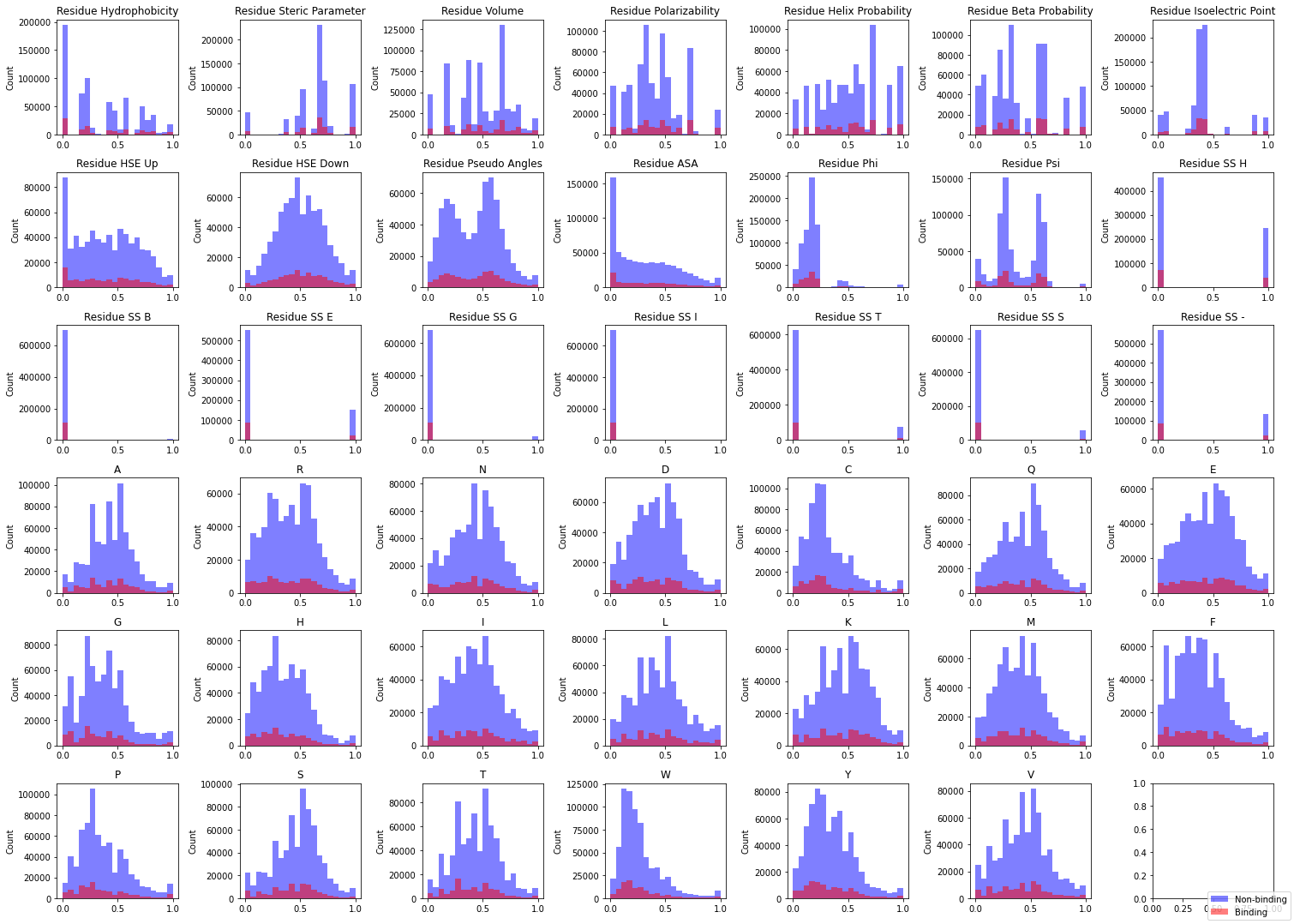 |
Figure 6. Distributions of 41 features found in PepBDB-ML by class.
The number of nonbinding residues far outnumbers binding residues, so it’s a little tricky to parse in this visualization — but feature distributions appear quite similar between samples.
Does this mean there isn’t a discernible difference between our samples? This could cause some issues if we plan on using this dataset for training classifiers. Let’s do some quick statistical analysis:
Statistical tests #
Small p-values for both the KS and U-test tell us there are statistically significant differences between the distributions of nonbinding and binding samples.
However, a lot of this is due to the difference in sizes of both samples. Looking at the effect size (Cohen’s d) we see these differences are quite small.
To truly parse any meaningful difference between the groups, we’ll have to leverage a machine learning model.
The Discussion: Key takeaways and learnings #
Developing this dataset was step 1 in trying to reimplement the code from the paper I mentioned at the start.
I (naively) assumed the data construction would be the easy part. In reality, the bulk of my time was spent wrangling the myriad of enrichment tools and databases, accounting for edge cases in all of them, and formatting.
But it paid off.
The CNN I trained on this data outperforms the original by 12 pts. on binding site detections (79%) — but I’ll save that for a future article.
Key takeaways:
A model is only as good as the data that it’s trained on. Garbage in, garbage out, as they say.
In any machine learning project, the hardest part is often data preprocessing.
Put effort into curating better, higher-quality datasets, and you’ll reap the rewards.
You can download the full dataset at my GitHub or my website. Running the data generation scripts can take some time, so ensure you have enough computing power or access to an HPC.
Let me know if you have any feedback on improving — thanks for reading!